En alternativ populasjonsgenetisk modell
(Fra Theistic Evolution; A Scientific, Philosophical and Theological Critique, kap. 16; Ola Hössjer, Ann K. Gauger og Colin R. Reeves)
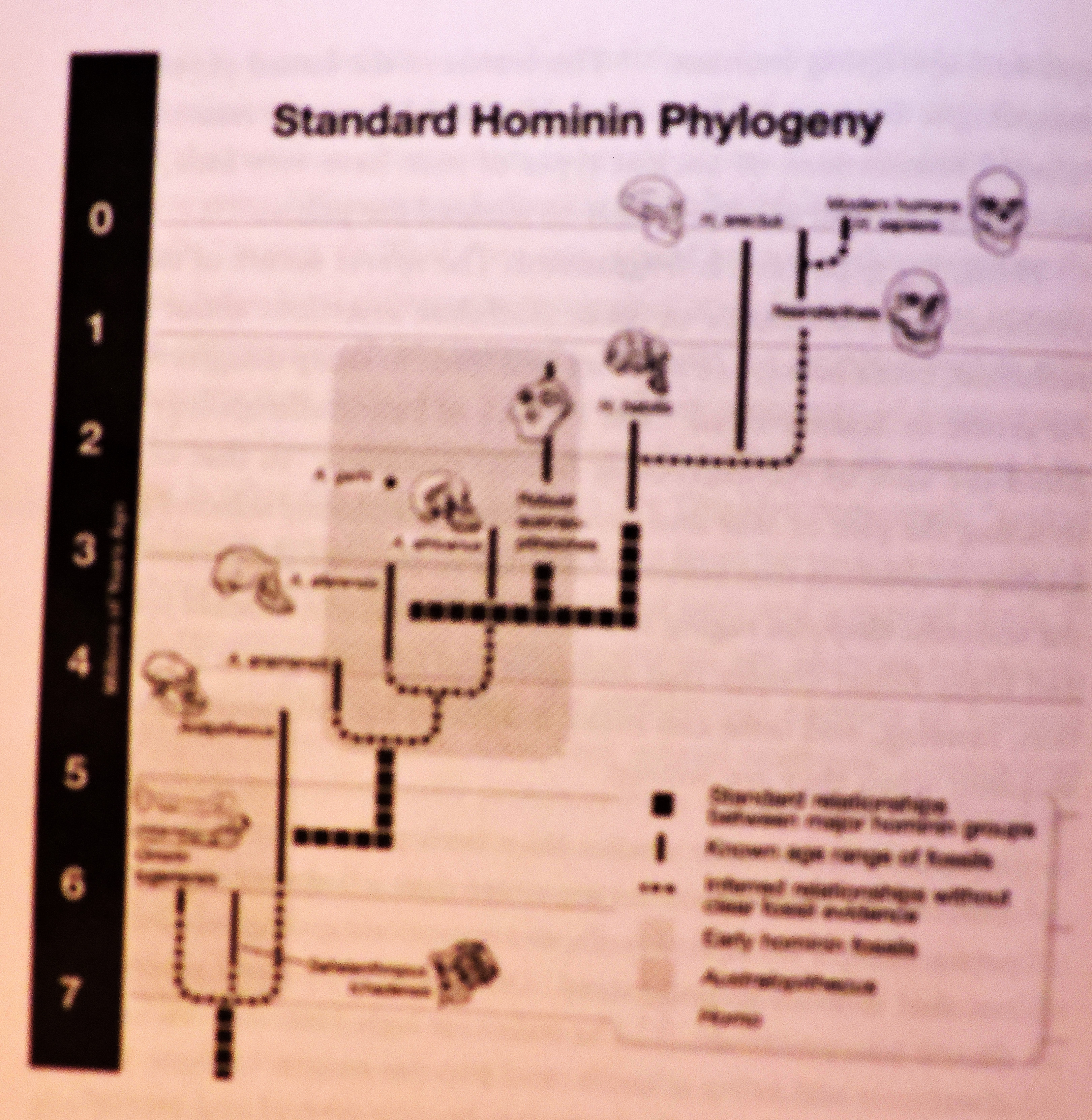
Bilde 1. Fra Fig. 14-1 (samme bok)
Oppsummering
Hva kan bli sagt om menneskets historie ut fra DNA-variasjoner blant oss i dag? Populasjonsgenetikere blir brukt i akademia for å slutte hvorvidt vi deler en felles avstamning med aper, at de fleste av våre menneskelige forfedre emigrerte fra Afrika for femti tusen år siden, at de mye mulig hadde en form for miks med Neanderthaler, Denisovanere og andre utdødde populasjoner og at den tidlige Homo-populasjonen ikke var mindre enn noen få tusen individer. Den benytter matematiske prinsipper for hvordan genetisk komposisjon til en populasjon endrer seg over tid, gjennom mutasjoner, genetisk drift og andre endrings-faktorer.
Dette kapitlet undersøker forutsetningene for denne teorien og finner at den er full av hull og svakheter. Vi argumenterer at en unik opprinnelsesmodell, der mennesket oppsto fra et enkelt par (2), synes å forklare dataene i det minste like godt, om ikke bedre. Til slutt foreslår de en alternativ simulerings tilnærmingsmåte (pkt.7), som kunne bli benyttet til å validere en slik modell.
Innledning: Vi har alle genetiske fingeravtrykk. Dess nærmere beslektet vi er, desto likere er våre fingeravtrykk. Genetiske data kan bli benyttet til en rekke ulike formål.. I dette kapitlet vil vi undersøke hva menneskelige genetiske data har å si om felles avstamning. I akademia er det rådende syn at menneskelige vesener er etterkommere av ape-lignende forfedre hvis antall aldri var mindre enn noen tusen individer på noe punkt i historien.
Men det er et annet mulig scenario, likevel, det med et opprinnelig første par. Dette scenariet har aldri blitt tilstrekkelig testet vitenskapelig (2017) ved å benytte metodene med populasjonsgenetikk. Gitt viktigheten til emnet er det nødvendig å grundig undersøke begge scenarier. Her beskrives en metode til å teste dem mot hverandre.
1. Populasjonsgenetikk
Populasjonsgenetikk er en disiplin som beskriver hvordan den genetiske makeup av mennesket endrer seg over tid. Den har flere anvendelser, men her vil vi bruke den som et verktøy for å sammenligne ulike scenarier av menneskets historie. Før vi starter med den kan det være nødvendig for noen å se på noen basisprinsipper for genetikk.
Genetisk informasjon ligger lagret i våre celler som 46 kromosomer, 23 fra hver av våre biologiske foreldre. 44 av disse er ikke-kjønnskromosomer (autosomale), og kommer i nesten  identiske (homologe) par. De resterende to kromosomer bestemmer vårt biologiske kjønn. Kvinner har to kopier av ett X-kromosom, ett fra hver forelder, mens menn har ett Y-kromosom fra faren, og ett X-kromosom fra moren. Det er også genetisk informasjon i mitokondriene, arvet fra vår biologiske mor.
identiske (homologe) par. De resterende to kromosomer bestemmer vårt biologiske kjønn. Kvinner har to kopier av ett X-kromosom, ett fra hver forelder, mens menn har ett Y-kromosom fra faren, og ett X-kromosom fra moren. Det er også genetisk informasjon i mitokondriene, arvet fra vår biologiske mor.
De 46 kromosomene i cellen og mitokondriene består av molekyler. Den genetiske informasjonen er lagret i disse molekylene som nukleotider linket sammen for å danne den dobbelt-dekkede DNA-heliksen (med vindeltrapp-form). Hver nukleotide, eller base, danner par med hverandre på stereotypt vis: A med T og C med G. (Se Fig. 16.1) Totalsummen med DNA i en av våre celler, kan bli tenkt på som en bok med 3 milliarder basepar av DNA. De fleste delene av DNA er de samme for alle mennesker, men de som varierer, kalles polymorfismer. Det er dem som gjør oss genetisk unike. En Enkel Nukleotid Polymorfisme (ENP) er den mest vanlige form for variasjon. For hvert basepar i DNA-molekylet, er konvensjonen bare å referere til én av dens to nukleotider, den som sitter på den 'kodende' siden.(Dette er den siden som involveres i protein-koding). Mindre enn 2% av den kodende siden av menneskelig DNA, koder faktisk for proteiner. Denne kodende delen består av et antall eksoner som først kodes (transkriberes) over til mRNA og så oversettes til proteiner. Denne nukleotiden eksisterer vanligvis i to varianter ved en ENP, for eks. C eller T, som også kalles de to allellene til ENP-en. Et typisk menneske-genom varierer litt mer enn 0,1% fra den menneskelige referanse-sekvensen (-som har det mest vanlige ENP i alle posisjoner).
Siden våre genom ikke er identiske, kan populasjons genetikk fortelle oss noe om menneskelig variasjon og historie. Vi kan studere hvor store de genetiske forskjellene er og indirekte noe om hvordan disse har endret seg gjennom historien. .. Populasjonsgenetikk var også ment som en metode for å forklare hvordan små endringer kunne føre til større endringer. Det ble altså i hovedsak grunnlagt for å støtte makroevolusjon og felles avstamning, basert på forestillingen at makroevolusjon bare var utvidelse av mikroevolusjon. Men i våre dager er det mulig å benytte den til det motsatte formål, å vise hvor usannsynlig makroevolusjon er. (10) Vi skal beskrive det mer detaljert i det videre.
2. Mekanismer for populasjonsendringer
Våre genom er omkastede bilder av våre forfedres genom. For å forstå hvordan historien har kastet om på våre forfedres DNA, trenger vi først å beskrive mekanismen som forårsaker at genetisk sammensetning til en populasjon endrer seg over tid. Disse mekanismene reflekterer populasjons-oppførsel (demografi) så vel som genetisk arv, og kan slik fortelle oss noe om en populasjons historie. De kan oppsummeres slik:
 Mutasjoner er endringer i DNA. Disse mutasjonene er typisk kopieringsfeil. De mutasjonene som er av interesse i populasjonsgenetikken er de som inntreffer når kjønnsceller dannes under en spesiell type celledeling -meiose: Disse halverer det genetiske materialet og reduserer antall kromosomer fra 46 til 23. Disse mutasjonene er viktige siden de sprer seg til alle cellene til avkommet etter befruktning. Anta f.eks. at en enkelt nukleotide blir endret fra A til G i en spermcelle som befrukter et egg. I det embryoet utvikler seg, vil alle celler arve den muterte allellen G fra faren, så vel som morens versjon, f.eks. A. De to versjonene av det nedarvede DNAet danner et par AG i alleler til det nye individet. Det individet vil i sin tur sende videre enten A eller G til hver av sine avkom, vanligvis tilfeldig. Mutasjoner forekommer også i sex-kromosomer og mitrokondriell DNA.
Mutasjoner er endringer i DNA. Disse mutasjonene er typisk kopieringsfeil. De mutasjonene som er av interesse i populasjonsgenetikken er de som inntreffer når kjønnsceller dannes under en spesiell type celledeling -meiose: Disse halverer det genetiske materialet og reduserer antall kromosomer fra 46 til 23. Disse mutasjonene er viktige siden de sprer seg til alle cellene til avkommet etter befruktning. Anta f.eks. at en enkelt nukleotide blir endret fra A til G i en spermcelle som befrukter et egg. I det embryoet utvikler seg, vil alle celler arve den muterte allellen G fra faren, så vel som morens versjon, f.eks. A. De to versjonene av det nedarvede DNAet danner et par AG i alleler til det nye individet. Det individet vil i sin tur sende videre enten A eller G til hver av sine avkom, vanligvis tilfeldig. Mutasjoner forekommer også i sex-kromosomer og mitrokondriell DNA.
Fig.2 Rekombinasjon: Her fremstår to ENP-er i samme ikke-kjønnskromosom, og anta at det bare er AC og TG kombinasjoner på disse posisjonene i populasjonen. Rekombinasjoner vil gradvis bryte opp denne A-C og T-G kombinasjonen. Det vil medføre at kromosomer med alle fire kombinasjoner (AC, AG, TG og GC) kan dannes. Grunnen er at en rekombinasjon mellom en foreldes to homologe kopier av kromosomet (på toppen) resulterer i skyfling av alleler når enten to spermceller eller to eggceller (bunnen) dannes. På figuren blir A og C alleler i foreldrens kromosom 1 rekombinert, slik at det nye kromsom 1 inneholder A og G i disse posisjonene, og det nye kromosom2 inneholder T og C.
Naturlig seleksjon er lik genetisk drift. Om foreldre med flere kopier av en spesiell allele, si A i en ENP, synes å være mer egnet (fit) enn de som har flere kopier av den andre allelen (si T) , vil frekvensen av A øke i neste generasjon, og evt. senere vil T forsvinne fra hele populasjonen (directional selection). Om derimot individer med en kopi hver av A og T (allele par AT), er mer egnet, så vil frekvensen av begge alleler tendere i å stabilisere seg over tid (balanserende seleksjon).
Rekombinasjon er en måte å danne mer variasjon mellom kromosomer ved å skyfle arrangement av alleler langs kromosomer. Det produseres ikke nytt DNA, men eksisterende DNA kombineres på nye måter. (Fig 2) Rekombinasjoner gjør variasjoner ved ulike deler av kromosomet mer uavhengig, i det de gradvis bryter opp sammenheng mellom alleler. Dette skjer f.eks. mellom A i den første ENP på kromosom 1 og C på den andre ENPen.
Kolonisering, isolering og migrasjon. Særlig i fortiden var mennesker mer eller mindre isolerte av avstand, mens de levde i nærhet av dem de paret seg med. Noen ganger dannes nye populasjoner (kolonisering), og noen ganger emigrerer menn og/eller kvinner over større avstander, for å finne sine partnere. Det er klart at migrering vil minke forskjellene mellom sub-populasjoner, mens isolasjon vil øke dem.
 3. Statistikk som beskriver genetisk variasjon i en populasjon.
3. Statistikk som beskriver genetisk variasjon i en populasjon.
Det finnes visse statistikker som beskriver nåværende genetisk variasjon i menneske-populasjonen. Nukleotide (allele) diversitet sier røft hvor mange ENPer det er i populasjonen generelt. Allele-frekvens spektre viser hvor mange sjeldne og felles alleler det er. Lenke-ulikevekt (Linkage Disequilibrium -LD) plott gir en beskrivelse av hvordan rekombinasjoner har påvirket variasjonene vi ser langs kromosomer i dag. Dess flere rekombinante begivenheter det fantes i fortiden, dess kortere er stykkene av DNA som er arvet sammenhengende. Det finnes også andre typer statistikk, som kan beskrive graden av ulikhet mellom subpopulasjoner.
Når populasjonsgenetikere forsøker å rekonstruere menneskelig historie fra genetiske data, kombinerer de ulike demografiske scenarierer (F.eks. populasjons -størrelser/endringer, kolonisering og migrasjon) med genetiske mekanismer til mutasjoner, rekombinasjoner, genetisk drift og seleksjon) for å se hvor godt disse scenariene rekonstruerer statistikken nevnt ovenfor. Det finnes høyst sofistikerte matematiske metoder for å gjøre dette, men det er likevel vanskelig å rekonstruere menneskelig historie. Hovedgrunnen til dette er mangel på data fra fortiden. Det er faktisk mulig å sekvensiere DNA fra gamle folkegrupper som neandertalene, og i senere år har denne forskningslinjen eksplodert. (14)
Til tross for dette, er fremdeles gammelt DNA så sjeldent at, i stor grad, vil genetiske analyser fortsette å støtte seg til DNA-prøver fra de mest nylige generasjonene. Med lite tilgjengelige historiske data, så er enhver rekonstruert genealogi, bare estimater med forutsetninger knyttet til seg. For å se hvordan fortiden har formet dagens genetiske variasjon, la oss betrakte allele diversitet. Det er en balanse mellom genetisk drift, mutasjoner, rekombinasjoner, seleksjon og migrasjoner. Genetisk drift minker diversiteten, siden noen alleler går tilfeldig tapt, desto raskere desto mindre populasjonen er.
Mutasjoner og rekombinasjoner vil på sin side øke ulikheten (diversiteten), siden enten nye alleler tilkommer populasjonen, eller gamle kombineres på ulike måter. Naturlig seleksjon kan enten minke eller øke allele-diversitet. Den vil avta, dersom alleler med høyere fitness tar over, eller om skadede alleler går tapt. Diversiteten vil øke dersom om koeksistente alleler har en selektiv fordel. Migrasjon og kolonisering skyfler rundt på eksisterende alleler mellom sub-populasjoner. Dess mer isolerte subpopulasjoner er, og dess mindre migrasjon det er mellom dem, desto større er diversiteten i populasjonen som helhet, siden ulike alleler tenderer i å ta over ulike sub-populasjoner. Innen hver subpopulasjon derimot, minker ulikhetene, grunnet isolasjon.
4. Alternative menneske-historier
 Det er to viktige, kontroversielle og sammenvevde aspekter i menneskelig historie: Alderen på vår opprinnelige befolkning og dens størrelse.
Det er to viktige, kontroversielle og sammenvevde aspekter i menneskelig historie: Alderen på vår opprinnelige befolkning og dens størrelse.
Alder: Hvordan kan vi benytte allele-ulikhet til å si noe om alder? På grunn av balansen mellom genetisk drift på den ene siden, og mutasjoner og rekombinasjoner på den andre. Populasjonsgenetikere argumenterer for at dess yngre en befolkning er, eller desto nyligere i fortiden den ble drastisk redusert (en såkalt flaskehals) desto mindre variasjon ser vi mellom de levende medlemmer av populasjonen i våre dager, fordi det har vært mindre tid for mutasjoner til å produsere nye varianter. Liten forskjell indikerer dermed en ung populasjon, eller en gammel som nylig gjennomgikk en trang flaskehals. Mer raffinerte konklusjoner kan trekkes, dersom vi også tar andre statistikker (allele-frekvens spektre, LD-plott og ulikhet innen subpopulasjoner) i betraktning. F.eks. LD-plott til en eldre populasjon indikerer at kortere DNA-sekvenser har blitt arvet sammen, siden det har vært mer tid til rekombinasjoner for å skyfle rundt kromosom-segmenter.
Størrelse: Basert på genetisk ulikhet og de andre typene statistikk, estimerer populasjonsgenetikere at vår populasjon aldri inneholdt mindre enn flere tusen individer, for å gjøre rede for nåværende diversitet, og at den inneholdt ca. 10 tusen på tiden vi hadde samme stamfar som sjimpansene, som de estimerer var ca. 6 millioner år siden. Begrunnelsen i dette argumentet er at forskjellen vi ser i menneskelig DNA, ikke bare er for stor til å kunne forklares ved et enkelt par, men den er også for stor til å forklares ved at en gammel populasjon som nylig gjennomgikk en flaskehals som var mye mindre enn noen få tusen individer.
Men det er imidlertid mulig at det første paret opprinnelig ble skapt iboende diversitet: Hvert individ med 22 par av autosome kromosomer samt enten XX-kromosom (kvinne) eller XY (for mann). Således ville det ha vært totalt fire kopier (to fra hver) av hvert ikke-kjønns kromosom. De fire kopiene kunne i utgangspunktet vært skapt unike, og gitt opphav til et veldig stor antall ENPer, som alle var til stede i første generasjon. Det ville holde stikk for alle de 22 settene med fire homologe kromosomer. På samme vis kunne de tre X-kromosomene, to for kvinne og en for mann, ha blitt skapt med diversitet. Noe diversitet fra mitokondrielt DNA er også mulig fra første generasjon. Siden kvinnen bærer hundrevis av mitokondrier som kunne ha vært ulike, kunne hun ha gitt videre noe av den diversiteten til sin datter.
Så vi har nå to konkurrerende hypoteser: felles avstamning av mennesker med andre arter, eller en unik opprinnelse for et første par, med skapt ulikhet. I neste avsnitt skal vi sammenstille disse to modellene: standardmodellen som formoder felles avstamning og den andre som forutsetter vi kom fra to første foreldre (unik opprinnelse). De tø modellene har ulik startforutsetninger, men følger samme regler for arv og populasjonsgenetikk i sine utarbeidelser.
 5. De konkurrerende modellene
5. De konkurrerende modellene
A. Felles avstamningsmodell
Den mest vidt aksepterte felles avstamnings-modellen for menneskeheten holder for at våre forfedre spredte seg ut fra sjimpansene for ca. 6 mill. år siden. Da utviklet en hominid art (Homo erectus) seg i Afrika. Den spredte seg ut til Europa og Asia for ca. 2 mill. år siden. Ulike utdødde arter antas å ha utviklet seg fra homo-erectus over de siste 500 -800 tusen år siden, inkludert Neandertalene i Europa og Denisovans i Asia. (Kap 14 i Theistic Evolution). Det er to hoved varianter innen dette rammeverket, for hvordan vår art homo sapiens ble til. 1. Ut av Afrika forflytnings-modell: I følge denne teorien utviklet moderne mennesker seg fra homo erectus i Afrika for 100-200 tusen år siden. De gikk gjennom en flaskehals, som reduserte bestanden til ca. 10 tusen individer eller mindre. En stor del av denne gruppen emigrerte fra Afrika for ca. 50 tusen år siden til Midt-østen, Europa, øst-Asia og Amerika, og erstattet gradvis opprinnelige arter. Etter å ha forlatt Afrika opplevde alle ikke-afrikanske populasjoner å ha opplevd mer nylige og langvarige flaskehalser før de startet å vokse i antall.
2. Multiregional evolusjon slår fast at våre stamfedre utviklet seg parallelt fra disse opprinnelige artene i flere ulike deler av verden, muligens med en Afrikansk dominans. Som en konsekvens må vi spore menneskelige linjer opp til 2 millioner år gamle, før de alle ender opp i Afrika.
Forflytningsmodellen har vært den mest populære avstamningsmodellen over flere tiår, men det er ingen bestemt grense mellom den og multiregional evolusjon. Et ut fra Afrika-scenario, med noe samvirke med opprinnelige populasjoner, er ikke for forskjellig fra en multiregional modell, med Afrikansk dominans. Over de siste få arene, har gammelt DNA blitt hentet fra Neandertal- og Denisovan-knokler i ulike deler av verden (18), og sml. med det til nåværende mennesker. Disse studiene viser at alle menneskelige populasjoner, unntatt sør for Sahara har mellom 1 og 2 % av neandertal-DNA. Faktisk virker det som så mye som 40% av deres DNA er funnet i det minste i noen menneskelige individer, som lever i dag. (19) Lavere nivåer (ca. 1% ) av Denisovan-opphav er funnet i Asia, Oceania og blant indianere. Dette har fått mange forskere til foreslå en hybrid av erstatnings og multiregional-modellen, der våre stamfedre kom fra Afrika, men også hadde noe samkvem med opprinnelige populasjoner. (20)
B. Unik opprinnelsesmodell
Ved unik opprinnelsesmodell mener vi en der menneskeheten oppsto fra ett enkelt par. Det er enda spørsmål om geografisk opprinnelse. Det er minst to versjoner av denne unike opprinnelsesmodellen, med ulikt geografisk opphav.
1. Afrikansk opphav. Det innebærer et scenario der det første paret levde i Afrika. Det har mange likheter med 'Ut fra Afrika' modellen, unntatt for den unike opprinnelses-formodningen. Den påfølgende migrasjonen ut fra Afrika, kunne være like for begge modeller. I neste avsnitt vil vi argumentere for at en unik opprinnelse med ut-fra-Afrika, typisk gir gamle estimater for menneskehetens alder.
2. Midt-østen opphav fastslår at den mest nylige felles stamfar til alle mennesker levde i Midt-østen. Den påfølgende migrasjonen fra Midt-østen til Europa, Asia, Amerika og Oseania, kunne være lik den fra ut-fra-Afrika og den unike opprinnelsesmodellen derfra (pkt. 1) Men denne skiller seg fra den første ved at den foreslår at Afrika ble kolonisert fra Midt-østen heller enn det motsatte. I neste avsnitt skal vi argumentere genetisk for at dette gir en mye yngre alder på menneskeheten.
 6. Sammenligning av modellene
6. Sammenligning av modellene
Det essensielle spørsmålet er hvilket av de ulike scenariene, enten felles avstamning eller unik opprinnelse, fra avsnitt 5 (foran), som de genetiske data støtter mest opp om. Forskjeller til andre arter: En hovedinnvending i forhold til modellen(e) for felles avstamning, er vansken med å håndtere de betydelige genetiske forskjellene mellom mennesker og andre arter. Det er faktisk forskningsartikler som forsøker å estimere en felles genealogi for mennesker, sjimpanser og gorillaer, ved å benytte de delene av genomet som viser mest likhet, men sammenlikninger av det slaget har begrenset rekkevidde, siden de fokuserer for lite på de regionene der artene skiller seg fra hverandre. (21) Modeller som sammenligner menneskelig DNA med det fra andre arter, skulle inkorporere vansken som mutasjoner og andre genomiske arrangement har i å bygge genomiske artsforskjeller, så vel som anatomiske og fysiologiske forskjeller. Andre studier som tar det i betraktning viser typisk at tiden det tar for de mutasjonene til å dukke opp og fikseres i populasjonen, er mye lengre enn at det kan forklares ved makroevolusjon. (22)
Variabilitet i menneskelige data. Hovedargumentet mot et unikt opphav, er at nukleotide diversiteten innen den menneskelige populasjonen synes for høy til å gjøre det mulig med et enkelt opprinnelsespar. Men det er ikke noe problem for ikke-kjønns kromosom og X-kromosomalt DNA, om det var en initielt skapt diversitet. Selv om det ikke er mulig å fremkalle skapt diversitet for Y-kromosomet, har nylige studier vist at dets diversitet uansett er mye mindre enn andre typer av DNA.
Genetiske data indikerer også at alle ikke-Afrikanske populasjoner, er ganske nært genetisk relatert. Dette forklares i 'Ut-av-Afrika' modellen ved en alvorlig og meget nylig flaskehals, i størrelsesorden 10 til 50 tusen år siden. Det er kanskje hovedgrunnen til at denne modellen er mer populær i dag enn multiregional evolusjon. Afrikanske populasjoner ser imidlertid eldre ut ved første syn, og det er også betydelige genetiske forskjeller mellom afrikanske grupper. (24) Tilhengere av 'ut-fra-Afrika' modellen fortolker flaskehalsen som at alle ikke-afrikanske mennesker emigrerte ut fra Afrika. De ikke-afrikanske populasjonene var lenge meget små, men så ekspanderte de hurtig og atskilte seg (25). Disse argumentene for 'ut-fra -Afrika' modellen synes overbevisende ved første øyekast. Men de kan også benyttes for den unike opprinnelsesmodellen, med et gammelt afrikansk opphav. Eneste forskjell er at det er ett opprinnelig par, som utgjorde det opprinnelige opphavet til alle folk som lever i dag, og som utvandret fra Afrika.
Hva med ett opprinnelig par fra Midt-østen? Er det unike opprinnelsen inkompatibel med data? Ikke nødvendigvis. Denne modellen krever at alderen på menneskeheten er mye yngre. Grunnen er at det opprinnelige paret evt. erstatter den langvarige flaskehalsen som inntraff etter immigrasjon til Midt-østen fra Afrika. (26) Og den genetiske diversiteten som er igjen etter denne flaskehals-populasjonen blir erstattet med skapt diversitet i det første paret. Det ville forklare den relativt store genetiske variansen vi finner blant mennesker i dag for ikke-kjønns og X-kromosom (-husk at diversiteten i Y-kromosomet er mye mindre.)
Blokk-struktur i DNA: En stor del av våre autosome (kromsom 1-22) og X-kromosomer har tilsynelatende blitt rekombinert i blokker av varierende lengde. (29) Mange av dem består av flere tusen nukleotider, men variasjonen i lengde er stor. Men selv om blokkene er lange, er det lite variasjon innen dem. Hver blokk kommer i bare noen få varianter -fire- for mange deler av genomet. Våre kromosomer er ulike mosaikker av disse blokk-variantene. Denne blokkstrukturen er forbløffende konsistent med en unik opprinnelseshypotese. Om det første paret oppsto med DNA-diversitet, så ville det ha vært fire ulike kopier av hvert autosomalt kromosom, to for hvert individ. Disse 4 kromosomer har siden blitt blandet med fortidige rekombinasjoner og i dag har hver av oss én mosaikk av de 4 opprinnelige kromosomer arvet fra våre fedre og en annen arvet fra vår mor.
 Innavl og genetisk entropi
Innavl og genetisk entropi
Det er velkjent at mange alleler vil gå tapt, grunnet genetisk drift, når en populasjon opplever en alvorlig flaskehals. Den langvarige konsekvensen er nedsatt evne for langvarig tilpasning til omgivelses-endringer, men det er også en mer akutt risiko for innavlsvikt når frekvensen av ressessiv uorden øker, i det flere avkom mottar en skadelig variant, fra begge foreldre, av et sykdomsfremkallende gen. Dess mindre populasjonen er og desto lengre tid det tar før størrelsen starter å øke, desto mer alvorlig er konsekvensen for populasjonens overlevelsesevne, slik at den til slutt kan dø ut. Innavlskrise er potensielt en vanskelighet for felles-avstamnings modellen. Nylige beregninger viser at modellen forutsier en meget trang flaskehals, i størrelsesorden av noen få tusen individer, og at det varte i minst tusen generasjoner. (35) Til tross for dette, mener man at overleverne av flaskehalsen skal ha bredt seg utover for å erobre resten av verden.
En unik opprinnelsesmodell unngår, i noen grad, problemet med innavlskrise, dersom den skapte diversiteten til det første paret bare hadde nøytrale varianter, mens skadelige mutasjoner i kjønnslinje-mutasjoner inntraff senere. Desto yngre populasjon, desto kortere tid har slike kjønnslinje-mutasjoner til å inntreffe, og desto mindre risiko for innavlskrise. Av lignende grunn vil en modell med en ung alder for menneskeheten ha en annen fordel: Den kan handtere situasjonen, at de mindre skadelige mutasjonene blir så mange, at naturlig seleksjon ikke kan flytte dem alle. I en gammel populasjon, i det mutasjonene akkumulerer over tid (teknisk betegnelse: genetisk entropi øker), så kan de potensielt ha meget skadelige effekter. (36) For gamle populasjoner kan enten antall lett skadde alleler øke, eller en flaskehals inntreffe, som flyttet noen av de skadelige variantene, men med den kostnad at andre ble spredd. Noe som igjen kan medføre en innavlskrise. (37)
Er opprinnelige (nå utdødde) populasjoner menneskelige eller ikke?
Som nevnt i avsnitt 5, er signifikante fragmenter av Neandertal og Denisovan -DNA funnet blant nålevende mennesker. Så forskning indikerer at noe seksuelt samkvem fant sted mellom gammeldagse populasjoner og den opprinnelige befolkning som formodentlig emigrerte ut fra Afrika. Dette er tenkt å ha funnet sted for minst 50 tusen år siden og senere. Det er velkjent at en genflyt mellom nært beslektede populasjoner er tjenlig for å unngå innavl, og oppnå genetisk variasjon. Men modellen for felles avstamning foreslår en splittelse av grener for mer enn 500 tusen år siden. (40) Det ville derfor være forunderlig om de to populasjonene ville være i stand til å ha fruktbart avkom, etter en så lang periode. Men selv om det hadde vært mulig, ville det være fornuftig å tro at avkommet hadde lav fitness (tilpasningsdyktighet), siden våre gammeldagse forfedre antagelig hadde akkumulert mange alleler som var ødeleggende for menneskeheten, før samkvemet fant sted.
I lys av dette, synes det som den store delen av opprinnelig DNA blant nåværende mennesker, er lettere forenlig med en unik opprinnelsesmodell, der neandertaler og denisovans er etterkommere etter det opprinnelige paret, og herav er våre fullt menneskelige slektninger.
Konklusjon: Vi har argumentert for at en unik opprinnelsesmodell (med enten en ung eller gammel menneskehet), med skapt diversitet skulle ha i det minste samme forklarende kraft for menneskelige genetiske data som den mest populære modellen i dag, med felles avstamning. Enhver modell må være i stand til å forklare store genetiske forskjeller mellom mennesker og andre arter, løse problemet med innavls-kriser, støtte levbarheten for blanding av moderne menneskelige og opprinnelige populasjoner, og gi grunner for at vårt DNA ligner en mosaikk av omkring fire grunnleggende genom-deler Konklusjonen er at den unike opprinnelses-modellen synes mest plausibel.
 7. Testing av modellene
7. Testing av modellene
Det kvalitative argumentet i avsnitt 5 ledet oss til å konkludere at den den unike opprinnelsesmodellen for menneskelig historie, virket plausibel. Det er av interesse å følge opp dette på en mer formell måte. Hovedideen er å simulere genetiske data fra hver av de foreslåtte modellene mange ganger, og så sammenligne hvor godt simulerte utdata passer til virkelige data. Sammenlikningen skulle inkludere nukleotide diversitet, allele-frekvens spekter, LD-plott og annen statistikk. Det er minst to ulike måter å fortsette med disse simuleringene.
Framlengs simulering. Den mest rett-fram måten å simulere genetiske data, er å starte med grunnlegger generasjonen og så fortsette framover i tid. (42) For hver simulerings-runde, tilordner man først genomet til det første par av mennesker. Demografiske og genetiske data blir så simulert en generasjon om gangen, ved å benytte arve-prinsippene i avsnitt 2.
Hovedfordelen ved framlengs simulering er dens store fleksibilitet. Nesten hver eneste modelltype for menneskelig historie kan bli simulert og validert med reelle data. Men metoden krever at DNA til alle mennesker blir simulert. I lys av størrelsen på den verdensvide menneskelige populasjon, er dette meget krevende å kompilere og krever lange eksekveringstider.
Baklengs simulering. Det er en annen, mye raskere, simulerings-algoritme. En mer delaljert beskrivelse av den kan bli funnet annensteds. (43) Vi arbeider nå på å implementere en modell basert på baklengs simulering. Hensikten er å validere den med relle data. Dette er et langtids-prosjekt, hvis resultat vi håper å kunne publisere annensteds. Ved å benytte denne tilnærmingen, er vår hensikt å demonstrere at en unik opprinnelses-modell er i stand til å replikere nåvårende menneskelig diversitet, like godt eller bedre enn den felles avstamnings-modellen. (45: Oppdatering på fremdriften til arbeidet ligger på uniqueoriginresearch.org -tilgjengelig med passord).
Det er vår hensikt med å teste denne modellen. Derfor, hvis mer enn en troverdig beretning av menneskelig avstamning kan forklare dataene, kan det ikke lenger bli hevdet at den felles avstamnings-modellen er avgjørende bevis for at det ikke kunne ha vært et første par. Dette var del av et mer omfattende argument (kap 14-16 i boka) for at det ikke er gitt at den tradisjonelle fortolkning for vår opprinnelse skal forkastes.
Til bokoversikten -her.
Referanser:
2: Bruk av termen opprinnelig par, vil uten tvil reise spørsmål om Adam og Eva i lesernes sinn. Forfatterne har sine egne syn på lesning av 1.Mos.1. Det som de vil ha fram her, er å vise at argumentet mot en historisk Adam og Eva gjort av noen vitenskapsfolk, ikke er begrunnet ut fra de vitenskapelige bevisene, og at det er en reell mulighet for et første par.
10: Faktisk har mange forskere som holder fast på felles avstamning for mennesker og sjimpanser, har nylig foreslått andre mekanismer enn makro-evolusjons drevne genetiske mekanismer for populasjons-endringer (pkt.2) og kap. 14 i boka Theistic Evolution. A .. Critique..
14: Mark Haber et al, "Ancient DNA and the Rewriting of Human History: Be Sparing with Occams Razor," Genome Biology 17, article 1 (2016): 1-8.
18: K. Prüfer et al; "The Complete Sequencing of a Neanderthal from the Altai Mountains," Nature 505, no.42 (2014):9; M. Meyer et al, "A High Coverage Genome Sequence from an Archaic Denisovan individual," Science 338 (2012):212-216
19: Svante Pääbo, "The Contribution of the Ancient Hominin Genomes from Siberia to Our Understanding of Human Evolution," Herald of the Russian Academy of Sciences 85, no.5 (2015):392-396
20: Haber et al; "Ancient DNA".
21: H. Kehrer-Sawatzki og D. Cooper, "Understanding of the Recent Evolution of the Human Genome: Insights from Human-Chimpanzee Genome Comparisons," Human mutation 28, no.2 (2007):99-130
22: John Sanford, W. Brewer et al, "The Waiting Time Problem in a Model Hominin Population," Theoretical Biology and Medical Modelling 12, art. 18 (2015):1-28
24: S.F. Schaffner, C. Foo et al, "Calibrating Coalescent Simulation of Human Genome Simulation,"Genome Research 15 (2005):1576-1583 m.fl.
25: 1000 Genomes Project Consortium, "Global Referance for Human Genetic Variation," 68-74. 26: Ibid.
29: To av de første publikasjons-artiklene om eksistensen av haplotype blokk-strukturer langs det menneskelige genom er:
M.J. Daily et al, "High-Resolution Haplotype Structure in the Human Genome," Nature Genetics 29(2001):229-232 og S.B. Gabriel et al., "The Structure of Haplotype Blocks in the Human Genome," Science 296(2002): 2225-2229
35: 1000 Genomes Project Consortium, "Global Referance for Human Genetic Variation," 68-74.
36: J. Sanford, Genetic Entropy and the Mystery of the Genome, 3.utg.(Waterloo, NY-FMS, 2008)
37: Dersom antall mindre skadede alleler er stort, så vil deres kumulative effekt også bli stor; Sanford, Genetic Entropy
40: Haber et al; "Ancient DNA".
42: J.C. Sanford, J. Baumgardner, W.Brewer et al, "Mendels Accountant: A Biologically Reasonable Forward-Time Population Genetic Program," Scalable Computin: Practise and Experience 8, no.2 (2007): 147-165.
43: Ola Hössjer, Ann Gauger og Colin Reeves, "Genetic Modelling of Human History Part2: A Unique Origin Algorithm," BIO-Complexity 2016, no.4 (2016):1-36
Oversettelse og bilder ved Asbjørn E. Lund