BIO-kompleksitet presenterer bedre modell enn 'Felles stamfar'
for å forklare naturens mønster
Av Brian Miller; 19. juli 2018 {Kursiv og understreking ved oversetter.}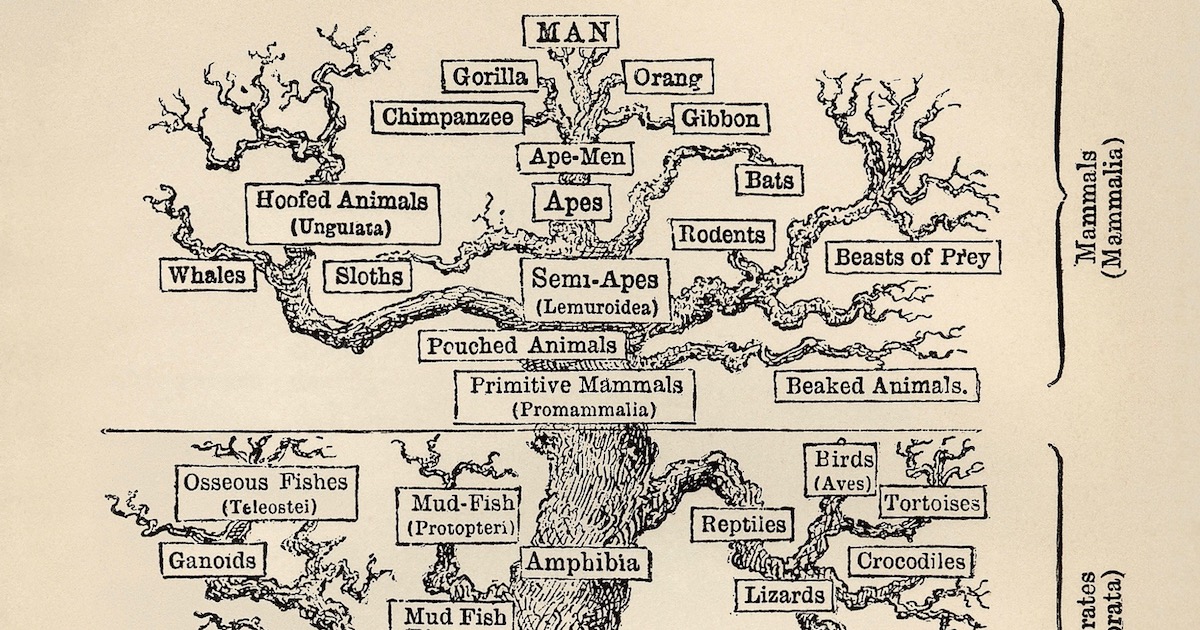
Bilde 1: Et tradisjonelt livets tre, via Wikimedia Commons.
En av de sentrale pilarene i den vanlige evolusjonære modellen er troen på at alle levende arter utviklet seg fra en felles stamfar gjennom et gradvis utvoksende 'livets tre'. Som et resultat antas mønsteret av likheter og forskjeller i arter i dag å passe inn i et trelignende mønster eller nøstet hierarki, hvor forgreningspunkter tilsvarer utseendet på nye egenskaper. For eksempel deler alle pattedyr visse egenskaper, som for eksempel melkeproduksjon, siden deres siste felles stamfar først utviklet disse egenskapene, og egenskapene løp gjennom hver utvoksende gren på treet. Dette ikoniske livets tre har blitt presentert for publikum som et av de sterkeste bevisene for utvikling av livet som skrider fram bare gjennom ikke-styrte, naturlige prosesser. Imidlertid har mange aspekter av denne historien blitt motsagt av store funn i løpet av de siste tiårene.
Av primær viktighet er at de første representanter fra de fleste større grupper av organismer oppstod plutselig i det fossile registeret -her uten identifiserbare sekvenser av mellomformer som fører tilbake til en felles stamfar med andre grupper. Like problematisk, innebærer ikke mønsteret av fysiske egenskaper og molekylære sekvenser i arter gjennom hele naturen, et konsistent evolusjonært tre. For eksempel er øynene til mennesker og åttearmet blekksprut ganske like, men de to gruppene antas bare å være fjernt relaterte. Ved å fremheve denne utfordringen, analyserte en eldre artikkel gjennom flere studier på tvers, prosentene av egenskaper for gitt grupper av arter, som passer konsistent med de best konstruerte kladogrammene (tilnærminger til et evolusjonært tre) -her.
Disse prosentene kjent som "konsistensindekser" ble deretter plottet på samme graf som de som ble avledet av tilfeldig generert data, og indeksene ble deretter justert for å fjerne effekten av tilfeldig støy. Gjennomsnittet for de justerte indeksene lå et sted rundt 0,35. Nyere forsøk på å konstruere kladogrammer for ulike grupper har ikke gått bedre, for eksempel med Euarchonta (gruppe inkludert i primater) og Therapsids (foreslåtte stamfar til pattedyr). Med andre ord passer omtrent to tredjedeler av alle dataene ikke til felles-avstamnings modellen.
Skuffende resultater, ad hoc-mekanismer
Disse skuffende resultatene har medført at evolusjonister har utviklet flere ad hoc-mekanismer for å forklare allestedsnærværende inkonsekvenser. Eksempler inkluderer Lateral GenOverføring (LGO), differensielt gentap og konvergent utvikling. Likevel har den brede appellen til LGT blitt seriøst betvilt -her. Og påstanden om at komplekse tilpasninger kan opptre uavhengig flere ganger (konvergent evolusjon) kollapser ved nøyere undersøkelser på grunn av usannsynligheten for at de fremkommer gjennom ukjente prosesser én eneste gang.
For eksempel antas øyne med linser å ha utviklet seg uavhengig flere ganger, men alle scenarier står overfor uoverstigelige barrierer når det gjelder motstridende selektivt press -her og nødvendige tidsskalaer -her. Enda mer slående involverte den påståtte konvergente utviklingen av ekkolokalisering i flaggermus og delfiner de samme sekvensmodifikasjoner i over 200 regioner av deres DNA -her. Imidlertid er tiden tilgjengelig for et terrestrisk pattedyr for å utvikle seg til et helt marint dyr utilstrekkelig til å frembringe til og med to nye koordinerte mutasjoner -her, og transformasjonen til hav-pattedyr krevde mange flere modifikasjoner -her. Til tross for disse utfordringene har evolusjonister opprettholdt at felles stamfar fortsatt er den beste forklaringen på dataene, siden det til en viss grad passer til et trelignende mønster. Denne påstanden står nå overfor en formidabel utfordring fra en nylig artikkel i tidsskriftet BIO-Complexity -her, av Winston Ewert. Han presenterer overbevisende et nytt rammeverk for 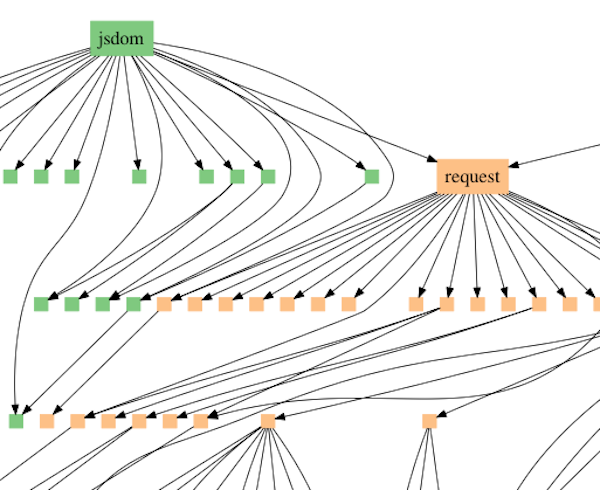 bedre å forklare naturens mønster.
bedre å forklare naturens mønster.
Et nytt rammeverk
Ewert modell tolker mønsteret av likheter i forskjellige grupper av arter som passer inn i det som refereres til i datavitenskap som avhengighetsgrafer. Nærmere bestemt skriver programmerere vanligvis ikke helt nye programmer helt fra grunnen av. I stedet gjenbruker de standardmoduler. Et eksempel er JavaScript-modulforespørselen, som laster ned filer fra Internett. Moduler har vanligvis tilgang til andre moduler, som danner et forgreningsnettverk av avhengighetsforhold (se figur 1). Java-script-forgrening.
Figur 1: Avhengighetsdiagram som illustrerer modulen jsdom som aksesserer modulforespørselen, som aksesserer andre moduler. Forespørselen er også aksessert av andre moduler på høyere nivå.
 Avhengighetsgrafer inkluderer ofte et nøstet hierarki som en del av sin struktur, men de inneholder også flere relasjoner som strekker seg utover et enkelt tre. Forskjellen kan sees ved å sammenligne det standard pattedyr-treet (figur 2) basert på felles avstamning , med en avhengighetsgraf avledet fra samme art (figur 3).
Avhengighetsgrafer inkluderer ofte et nøstet hierarki som en del av sin struktur, men de inneholder også flere relasjoner som strekker seg utover et enkelt tre. Forskjellen kan sees ved å sammenligne det standard pattedyr-treet (figur 2) basert på felles avstamning , med en avhengighetsgraf avledet fra samme art (figur 3).
Figur 2: Livets liv for utvalgte pattedyrarter. Hvert sett av arter på et gitt nivå i det nestede hierarkiet har bare den siste felles stamfar. Pattedyr-tre.
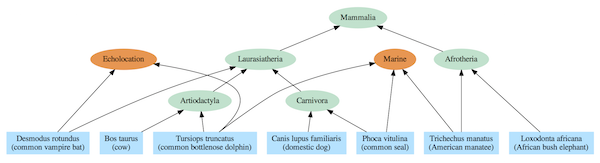
Figur 3: Avhengighetsgraf for utvalgte pattedyrarter. En enkelt art kan avhenge av flere moduler. Pattedyr-avhengighetstre
Den evolusjonære tremodellen krever, i hvert fall for komplisert liv, at alle arter knytter seg tilbake til en enkelt Siste Felles Stamfar (SFS)med andre arter i samme gruppe (kladogram). For eksempel er elefanter og sjøpattedyr begge medlemmer av "afrodyr"-kladen (monofyletisk gruppe), så de er avbildet som å ha en SFS som var den gruppens første representant. Og hver SFS kobler tilbake til en enkelt SFS for alle medlemmer av grener (klader) på høyere nivåer i hierarkiet. For eksempel deler SFS til klovdyr og kjøtteter-grenene en enkelt SFS med alle medlemmer av placenta-dyr grenen. Og alle pattedyr arter knytter seg tilbake til en enkelt SFS som er roten til pattedyr-treet.
Den tilsvarende avhengighetsgrafen inkluderer det samme nestede hierarkiet som det påståtte evolusjonære treet, men relasjonene tolkes ikke i forhold til SFS, men ved hjelp av delte moduler. For eksempel bruker elefanter og sjø-pattedyr begge Afrotheria-modulen, og alle pattedyr benytter pattedyr-modulen. Likevel, i sterk kontrast til den vanlige stamfar- modellen, bruker sjøpattedyr, sel og delfiner alle havdyr-modulene som er inkongruent med det evolusjonære treet. På samme måte bruker flaggermus og delfiner begge ekkolokasjonsmodulen, mens de ikke er nært beslektet i den felles avstamningsmodellen.
Sammenligning av to modeller
Ewert sammenlignet prediktiv kraft av avhengighetsgrafen og vanlige stamfarmodeller ved å analysere fordelingen av genfamilier i forskjellige samlinger av arter hentet fra ni forskjellige databaser. For hver database ble et sett gener brukt av flere arter, identifisert som en modul som disse artene avhenger av. Og et sett med gener som finnes i flere større moduler ble identifisert som en distinkt modul som de større modulene avhenger av. En optimaliseringsrutine ble brukt til å konstruere en tilnærming til den beste avhengighetsgrafen, og den grafen ble sammenlignet med livets tre som presenteres av NCBI-databasen hierarkiet. Den beste passform til dataene ble da bestemt mellom de to representasjonene ved hjelp av Bayesisk modellvalg.
 Avhengighets-graf modellen gjør flere forutsigelser som er i direkte motstrid til felles avstamnings-modellen:
Avhengighets-graf modellen gjør flere forutsigelser som er i direkte motstrid til felles avstamnings-modellen:
*Biologiske data skulle passe en avhengighetsgraf bedre enn et tre.
*Data produsert fra en prosess dominert av felles nedstigning eller forgrening bør passe et tre bedre enn en avhengighetsgraf.
*Avledede grafer for biologiske data bør inneholde mange flere ikke-taksonomiske moduler med mange flere gener, enn avhengighetsgrafer utledet fra data, kjent for å ha blitt produsert fra felles avstamning.
*Programvare bør passe en avhengighetsgraf bedre enn et tre, men et tre bedre enn en null-modell. En nullmodell tilsvarer et mønster som ikke-eksisterer for gjenbruk av genfamilier på tvers av arter.
Ewerts analyse stadfestet alle disse prognosene med høyt statistisk tillit for alle databaser. Derfor indikerer denne innledende studien at avhengighets-grafmodellen i stor grad overgår den felles avstamningsmodellen, i å forstå naturens mønster.
Som en konsekvens blir alle påståtte evolusjonære trær og sekvenser sterkt mistenkte, inkludert slike ikoner som hval- og menneske- serien. For de er basert på likheter av egenskaper mellom arter, og likheter er en upålitelig indikator for felles avstamning som antydet av trærnes typisk lavtilpassede konsistens-indekser. I stedet ser likheter ut til å være et resultat av en designers gjenbruk av designmoduler, i ulike arter for å møte felles mål.
Ewerts artikkel representerer bare det første trinnet i evaluering og utvikling av hans rammeverk. Likevel kan betydningen av denne undersøkelsen ikke overvurderes. Avhengighets- grafmodellen forklarer hvorfor delmengder av de biologiske data-formene bare grovt passer til et tremønster, og hvorfor så mye av dataene er inkongruente. Det gir også klare forutsigelser om resultatene av fremtidige studier om fordelingen på tvers av arters både fysiske egenskaper og likheter i molekylære data. Til slutt bør det føre til et robust og innovativt forskningsprogram, basert på det intelligent-design rammeverket.
Oversettelse og bilder ved Asbjørn E. Lund